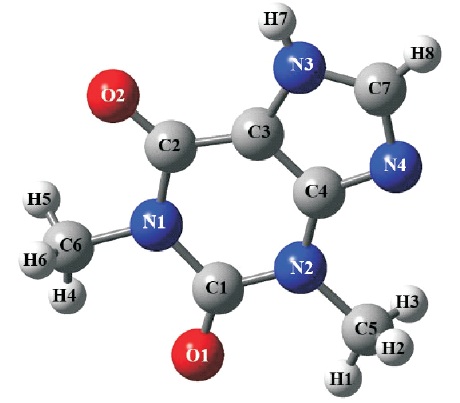
Figure 1. Single Molecular Structure of C7 H8 N4 O2
In this work, the Density Functional Theory (DFT) quantum mechanics computational method has been conducted on Theophylline molecular system. DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** level of theories were implemented in the whole calculations. Theophylline (TP) compound chosen has the chemical formula of C7H8N4O2 and lattice parameters of a=24.612 Å, b=3.8302 Å, c=8.5010 Å and α=β=γ=90°. Geometry optimization calculation has been performed to obtain the equilibrium structure of C7H8N4O2 molecular system. Moreover, the optimized geometry structure has been used to calculate the electronic properties (i.e., total energies, frontier molecular orbital energies, atomic charges, and others) of the studied molecular system. The calculated total energies obtained from DFT/B3LYP/6-31G method is -17438.43 eV, while from DFT/B3LYP/6-31G** method is -17444.53 eV. The HOMO-LUMO energy gaps of C7H8N4O2 molecular system are 5.00 eV and 5.12 eV respectively for DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** level of calculations. The molecular electrostatic potential (MEP) mapped plots obtained from both B3LYP/6-31G and B3LYP/6-31G** methods presented that the regions around the atoms of O1 and O2 possess the negative electrostatic potentials, while the positive electrostatic potential falls on the region of H7 atom. In addition, Fourier Transform Infrared (FT-IR) calculation is also included in this study. All the findings of vibrational frequencies show no surprising feature and has been in accordance with the recorded data.
The vigorous development in computer technology and further improvement in efficient computational method had brought revolutionary enrichment in quantum mechanics computational study. Density Functional Theory (DFT), as one of the quantum mechanics computational methods, gained its popularity over several decades in the investigation of electronic properties and vibrational spectral on atoms, molecules and condensed matter (Kirste, 2016; Ramya et al., 2013). Density Functional Theory shows better computational performance than Hatree- Fock method as it considers both exchange and correlation effects in electrons. Density Functional Theory has been able to provide more accurate data with reasonable computational time and cost (Zainuri et al., 2017). In this work, Density Functional Theory computational method has been employed to study the geometry structural, electronic properties and vibrational spectral of Theophylline (TP) drug compound. Polymorphism refers to the ability of a solid compound to exist in multiple crystalline structures that differ in crystal lattices or conformations (Raza et al., 2014). Each polymorphic form has different physiochemical properties, such as solubility, stability, melting point, hardness, and so on (Barnes, 2013). In pharmaceutical industry, the study and investigation on polymorphism phenomena in drug compound is significant since it affects the bio-availability and therapeutic performances of a drug. Therefore, both the experimental and theoretical studies on the characteristics of polymorphic drug compounds have become an essential and potent part in the drug development industry nowadays.
The title compound, 1,3-dimethylxanthine or commonly known as theophylline, can be easily found in cocoa bean and tea leaf (Barnes, 2013). Similarly other xanthine based compounds (i.e. theobromine and caffeine), theophylline effectively acts as phosphodiesterase (PDE) inhibitors (Barnes, 2013). Moreover, theophylline has the capability to relax the muscle, and acts as bronchodilator agent. To date, theophylline possesses outstanding therapeutic effect among the xanthine based drug compounds in the treatment of asthma and chronic obstructive pulmonary disease (COPD) (Barnes, 2013). In 2013, Barnes presented that the higher dose of theophylline drug was required during the treatment due to its low solubility and low bioavailability traits (Barnes, 2013). Theophylline has small therapeutic index and high toxicity to human body. Higher concentration of theophylline will cause several side effects, such as anxiety, diarrhoea, and heart palpitation. Theophylline is one of the active pharmaceutical ingredients (API) drug, which has the ability to form several polymorphic forms. Although theophylline drug has been discovered and applied in medical field a number of years ago, many researches still put effort in identifying and developing the polymorphic forms of theophylline to improve its pharmaceutical performances. For example, a monoclinic form of theophylline (C7 H8 N4 O2 ) compound with lattice parameters of a=7.8935 Å, b=12.9087 Å, c=15.9055 Å and β=104.214° has been discovered and reported by Zhang and Fischer in 2011 (Zhang & Fischer, 2011). In 2012, Fucke et al. presented four monoclinic and orthorhombic hydrated polymorphic forms of theophylline (Fucke et al., 2012). Moreover, Kakkar et al. (2018), studied the characteristics of polymorphic forms of theophylline experimentally through the formation of theophylline cocrystal with six different solvents, such as picolinamide, acetazolamide, and others. However, based on our literature studies, there have been no theoretical and computational studies on the electronic structure investigations of theophylline compound. In order to get a better understanding on the geometric and electronic structures of theophylline molecular system, a Density Functional Theory (DFT) computational method has been implemented. With the lattice parameters of a=24.612 Å, b=3.8302 Å, c=8.5010 Å and α/β/γ = 90°, an orthorhombic form of theophylline, which discovered and presented by Ebisuzaki et al. (1997) was chosen as local environment in this calculation.
Gaussian 09 software package (Frisch et al., 2019) has been chosen to perform all the DFT calculations through personal computer. The Crystallographic Information File (CIF) of theophylline compound has been obtained from the Cambridge Crystallographic Data Centre (CCDC) with the deposition number of 128707 (Ebisuzaki et al., 1997). A single molecule of theophylline compound has been extracted from the unit cell via GaussView 05 software (Frisch et al., 2019) and acts as the host environment for the entire calculations in this study. Geometry optimization calculations were performed using Becke's Three Lee- Yang-Parr (B3LYP) hybrid functional together with 6-31G and 6-31G** basis sets to obtain the equilibrium structures of C7 H8 N4 O2 . Figure 1 illustrates the single molecular structure of title compound used in this work. The optimized geometry structures were used to determine the geometrical parameters (i.e., bond distances, bond angles, and dihedral angles) and electronic properties (i.e., total energies, frontier molecular energies, dipole moment, and so on). In addition, the plots of molecular electrostatic potentials (MEPs) and frontier molecular orbitals (FMOs) were also presented in this study. Qualitative analysis has been carried out using Multifwn software (Lu & Chen, 2012). The formatted checkpoint file (.fchk) generated by Gaussain09 software acted as the main input source and the calculated global surface maxima and minima for C H N O molecular system were obtained.
Figure 1. Single Molecular Structure of C7 H8 N4 O2
Table 1 shows the optimized geometrical parameters of C7 H8 N4 O2 compound. Both of DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** methods show good agreement with the experimental data. From the table, the computed results clearly explains that the largest percentage difference between the computational and experimental bond distances is 3.42%. This is contributed by C7 –N3 bond distance and followed by the bond distances of C2 –O2 (3.17%) and C1 –N2 (3.06%). In the case of bond angle, the angles of O1 –C1 –N1 , C1 –N1 –C2 , C2 –C3 –C4 , and C4 –C3 –N3 are determined to be 1.43%, 0.46%, 1.14%, and 0.51% different from the experimental values. Moreover, with the corresponding percentage difference of 2.04%, the dihedral angle of C1 –N2 –C4 –C3 is contributed to be the largest percentage difference among all dihedral angles in the molecular system. Overall, all computed DFT values show slight difference with those of experimental findings due to the fact that experiment is recorded under bulk and solid state phase, while computational calculation is performed under gases phase (Ramya et al., 2013).
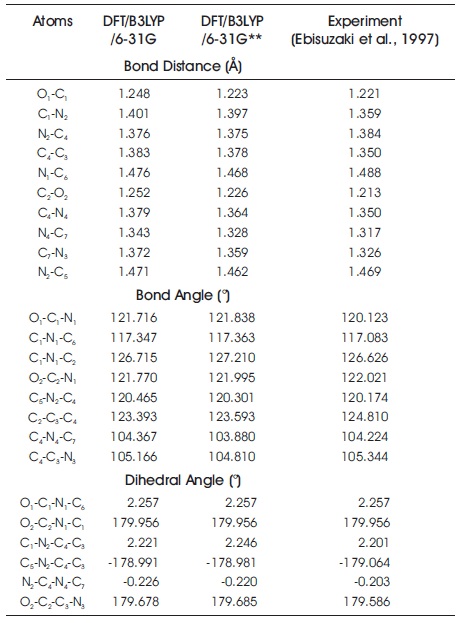
Table 1. Experimental and Calculated Geometrical Structures (i.e., bond distances, bond angles, and dihedral angles) of C7 H8 N4 O2 Molecular System.
Table 2 summarizes the computed electronic properties of C7 H8 N4 O2 molecular system. All computed results obtained from DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** calculations show good agreement with each other in this work. For example, the total energy and dipole moment of C7 H8 N4 O2 molecular system are -17438.433 eV and 3.622 Debye for DFT/B3LYP/6-31G method, while the method of DFT/B3LYP/6- 31G** possess -17444.527 eV and 3.462 Debye. In recent years, frontier molecular energies are immensely useful to identify and explain the chemical reactivity, kinetic stability, hardness, softness, and conductivity of studied system (Bhuvaneswari et al., 2018; Shafieyoon et al., 2019). HOMO known as highest occupied molecular orbital, is the outermost orbital that contain electrons and tend to give electron away. LUMO known as lowest unoccupied molecular orbital, is the innermost orbital that contain free places and act as electron acceptor (Demircioğlu et al., 2015). The HOMO energy indicates the electrons donating ability of C7 H8 N4 O2 molecular system, while LUMO energy determines the electrons accepting ability (Bouchoucha et al., 2018). The calculated energies of HOMO, LUMO, and HOMO-LUMO for B3LYP/6-31G calculation are -6.284 eV, - 1.285 eV, and 5.000 eV, respectively, whereas the energy values of -6.084 eV, -0.965 eV and 5.118 eV are determined for B3LYP/6-31G**. The HOMO-LUMO energy gap obtained from both calculations are approximately same with the findings (Al Shaabani et al., 2016; Salihović et al., 2014). Figure 2 illustrates the HOMO and LUMO surface mapped plots for both B3LYP/6-31G and B3LYP/6-31G** level of theories. The red colour lobe region represents the positive phase and the green colour lobe region represents the negative phase (Prabhaharam et al., 2015). The observation of figures shows the identical HOMO and LUMO surface plots, where the HOMO and LUMO electron distributions are mainly localised on entire theophylline molecule except the methyl group. In this studied system, the major contribution of HOMO is from the py orbital of O1 , C3 , C4 , N2 , O2 , N3 , and C7 . These atoms have better capability to donate the electrons in the molecular system (Prabhaharam et al., 2015). While for the case of LUMO, the distribution of electron density is mainly contributed by the py orbital of N1 , C2 , C3 , C4 , N2 , O2 , N3 , and C7 atoms, which have stronger capability to receive the electrons in the title molecule (Ramya et al., 2013). In this work, the Ionization Energy (IE) and Electron Affinity (EA) of C7 H8 N4 O2 molecular system can be obtained since the studied system possesses the negative sign of HOMO and LUMO energies. For DFT/B3LYP/6-31G level of theory, the ionization energy and electron affinity are found to be 6.284 eV and 1.285 eV. For DFT/B3LYP/6-31G** method, 6.084 eV and 0.965 eV corresponds to ionization energy and electron affinity, respectively. Moreover, the electro negativity (χ), hardness (η), softness (Ѕ), and chemical potential (μ) of studied molecular system can be predicted using the equations presented by Bhuvaneswari et al. (2018) and Shafieyoon et al. (2019). In this work, the electronegativity (χ), hardness (η), softness (Ѕ), and chemical potential (μ) are obtained with DFT as follows.
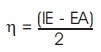
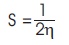
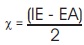
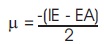
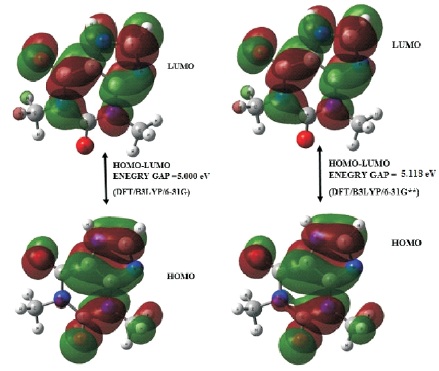
Figure 2. Frontier Molecular Orbitals of C7 H8 N4 O2 Molecular System Using DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** Levels of Theories
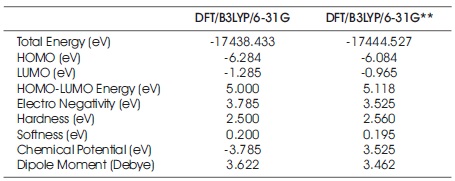
Table 2. Electronic Properties of C7 H8 N4 O2 Molecular System
In the case of B3LYP/6-31G, the global hardness, softness, electronegativity, and chemical potential are 2.500 eV, 0.200 eV, 3.785 eV, and -3.785 eV, while the values of 2.560 eV, 0.195 eV, 3.525 eV, and -3.525 eV are presented for B3LYP/6-31G** calculation. From the Table 2, it can also be found that the dipole moments of C7 H8 N4 O2 compound are 3.622 Debye and 3.462 Debye, respectively at DFT/B3LYP level of theory by employing the different basic sets of 6- 31G and 6-31G**. Table 3 tabulates the atomic charge distribution of the title compound based on the schemes of Mulliken Population Analysis (MPA) and Natural Population Analysis (NPA). According to the Table 3, there are no suspicious findings in MPA and NPA data using DFT/B3LYP/6- 31G and DFT/B3LYP/6-31G** methods. Both results of MPA and NPA obtained from B3LYP/6-31G and B3LYP/6-31G** calculations clearly noted that the highest positive charge falls on the atom of C1 . Overall, all the oxygen and nitrogen atoms possess the negative charge values in the studied molecular system. Also, the hydrogen atoms are found to have positive charge values. The molecular electrostatic potential (MEP) mapped plot of C7 H8 N4 O2 compound for both calculations are illustrated in Figure 3, respectively.
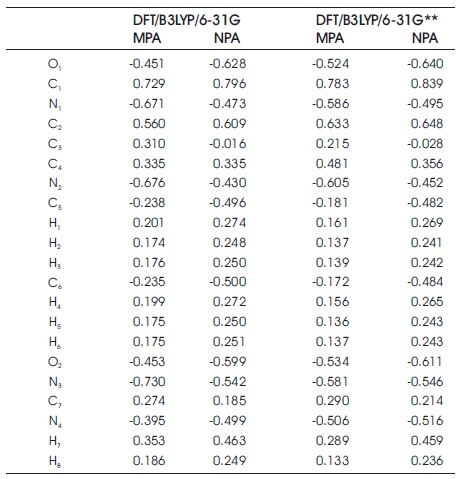
Table 3. Distributions of Atomic Charges of C7 H8 N4 O2 Molecular System Using the Schemes MPA and NPA
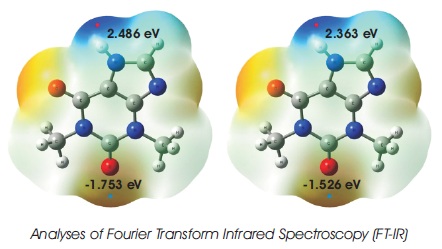
Figure 3. Molecular Electrostatic Potential (MEP) Surface Mapped Plots of C7 H8 N4 O2 Obtained from DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** Calculations
The surface diagrams of MEPs are immensely useful to understand the net electrostatic effect and reactive behaviour of C7 H8 N4 O2 compound in this work. From the Figure 3, it clearly noted that both O1 and O2 atoms are surrounded by red colour region, while H7 atoms are situated in blue colour region. These indicate that the region around the atoms of O1 and O2 possess the highest negative electrostatic potential, and will undergo electrophilic reactions in this study, whereas the atom of H7 will undergo the nucleophilic reaction and have highest positive electrostatic potential (Demircioğlu et al., 2018). In addition, the global surface maximum (blue dot) and global surface minimum (red dot) of C7 H8 N4 O2 molecular system are also presented in Figure 3. For DFT/B3LYP/6-31G calculation, the global surface minimum is located on the region around O1 with the corresponding potential energy value of -1.753 eV. The region around the atom of H7 is assigned to global surface maximum with the potential energy of 2.486 eV. In the case of B3LYP/6-31G** method, the global surface minimum and maximum are focused on the atom of O2 and H7 , with the predicted potential energies of -1.526 eV and 2.363 eV.
Table 4 summarizes all computed Fourier Transform Infrared Spectroscopy (FT-IR) results (with scaled and unscaled) of C7 H8 N4 O2 molecular system. In FT-IR frequencies assignment, it is recommended to employ the usage of scale factor in order to obtain more reliable data, since the vibrational frequencies calculation always ignore the anharmonicity of the real system (Rahmani et al., 2018; Shafieyoon et al., 2019). The scale factor of 0.962 and 0.961 are used for B3LYP/6-31G and B3LYP/6-31G** calculations in this study. For C7 H8 N4 O2 molecular system, the band regions of 3666 cm-1 (unscaled) – 3675 cm-1 (unscaled), and 3523 cm-1 (scaled) – 3535 cm-1 (scaled) are assigned to secondary amine N-H stretching vibrational mode. Moreover, the frequencies of 3070 cm (unscaled) – 3322 cm-1 (unscaled), and 2950 cm-1 (scaled) – 3196 cm-1 (scaled) are corresponded to C-H stretching vibration mode in this work. The C=O stretching vibration behaviour falls in the band regions of 1638 cm-1 (unscaled) – 1798 cm-1 (unscaled), and 1576 cm-1 (scaled) – 1727 cm-1 (scaled). In addition, the data from the table also indicates that the regions of 1064 cm-1 (unscaled) – 1263 cm-1 (unscaled), and 1023 cm-1 (scaled) – 1214 cm-1 (scaled) are corresponded to the aromatic ring C-N stretching vibration mode. The aromatic C=C bending vibration is observed on the frequencies of 735 cm-1 (unscaled) – 774 cm-1 (unscaled), and 707 cm-1 (scaled) – 744 cm-1 (scaled). Overall, all the computed vibrational frequencies obtained from both B3LYP/6-31G and B3LYP/6-31G** calculations are in accordance with the recorded spectral (Novena et al., 2017).
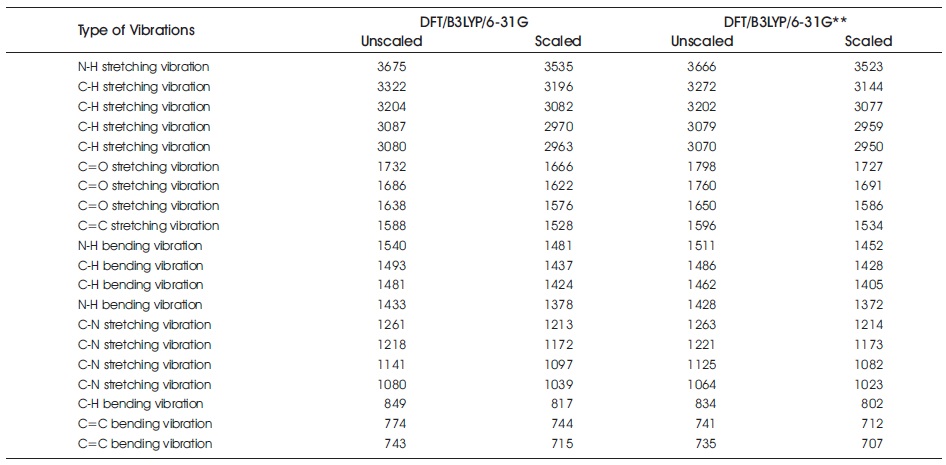
Table 4. Computed FT-IR Using DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** Methods
In this study, the geometric and electronic properties of C7 H8 N4 O2 molecular system were obtained using DFT computational method with the help of Gaussian09, GaussView05, and Multiwfn software packages. All optimized geometrical parameters show satisfactory agreement with the measurement presented by Ebisuzaki et al. (1997). The computed total energies are -17438.433 eV and -17444.527 eV, respectively for DFT/B3LYP/6-31G and DFT/B3LYP/6-31G level of calculations. Also, the dipole moment and HOMO-LUMO energy band gap obtained from B3LYP/6-31G method are 3.622 Debye and 5.000 eV, whereas the values of 3.462 Debye and 5.118 eV were predicted from the method of B3LYP/6-31G**. In the case of FT-IR frequency analysis, the findings of band assignments (unscaled and scaled) were matched with the functional group existing in the studied theophylline compound. However, all computed findings whether geometric or electronic structures in this study, provided useful fundamental knowledge of theophylline at the microscopic level. In addition, all the computed data using DFT/B3LYP/6-31G and DFT/B3LYP/6-31G** methods can act as references for other researchers when they deal with theophylline polymorphs in the future.