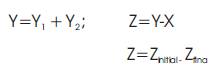
Although no single drug has been designed solely by computer techniques, the contribution of these methods to drug discovery is no longer a matter of dispute. All the world's major pharmaceutical and biotechnology companies use computational design tools. At their lowest level the contributions represent the replacement of crude mechanical models by displays of structure which are a much more accurate reflection of molecular reality, capable of demonstrating motion and solvent effects. Beyond this, theoretical calculations permit the computation of binding free energies and other relevant molecular properties. The theoretical tools include empirical molecular mechanics, quantum mechanics and, more recently, statistical mechanics.
This paper is concerned with the modeling of a new drug for heart disorders making 3-hydroxy-3-methylglutarylcoenzyme A reductase (HMG CoA reductase) as a target and Atorvastatin, as the drug used. This drug is modified structurally and its derivatives obtained as such are tested for their affinities in binding with the protein. Objectives of this paper are to optimize the structures of the drug derivatives, to calculate the energies of the optimized derivated structures and optimized protein-ligand structures, to dock the ligands (derivatives) with the protein structure and calculate the binding affinities of each molecule, to compare the binding energies and identify the best lead compound with maximum stability.
The vast majority of drugs are small molecules designed to bind, interact, and modulate the activity of specific biological receptors. Receptors are proteins that bind and interact with other molecules to perform the numerous functions required for the maintenance of life. They include an immense array of cell-surface receptors ( hormone receptors, cell - signaling receptors, neurotransmitter receptors, etc.), enzymes, and other functional proteins. Due to genetic abnormalities, physiologic stressors, or some combination thereof, the function of specific receptors and enzymes may become altered to the point that our well-being is diminished. The role of drugs is to correct the functioning of these receptors to remedy the resulting medical condition.
Drug design is the approach of finding drugs by design, based on their biological targets. Typically a drug target is a key molecule involved in a particular metabolic or signalling pathway that is specific to a disease condition or pathology, or to the infectivity or survival of a microbial pathogen.
Computer-Aided Drug Design (CADD), represents more recent applications of computers as tools in the drug design process. CADD uses computational chemistry to discover, enhance, or study drugs and related biologically active molecules. Methods used can include simple molecular modeling, using molecular mechanics, molecular dynamics, semi-empirical quantum chemistry methods, abinitio quantum chemistry methods and density functional theory. The purpose is to reduce the number of targets for a good drug that have to be subjected to expensive and time-consuming synthesis and trailing.
Heart disease is an umbrella term for a number of different diseases affecting the heart. As of 2008, it is the leading cause of death in the world especially in the United States, England, Canada, Wales and India [3]. Cardiovascular disease refers to the class of diseases that involve the heart or blood vessels (arteries and veins).
The drugs that target the Hmg coA reductase are used to cure heart diseases. The enzyme 3-hydroxy-3- methylglutaryl coenzyme A (HMG-CoA) reductase catalyzes the conversion of HMG-CoA to mevalonate, a four-electron oxidoreduction that is the rate-limiting step in the synthesis of cholesterol and other isoprenoids. The enzyme is found in eukaryotes and prokaryotes; and phylogenetic analysis has revealed two classes of HMGCoA reductase, the Class I enzymes of eukaryotes and some archaea and the Class II enzymes of eubacteria and certain other archaea [4,5]. Three-dimensional structures of the catalytic domain of HMG-CoA reductases from humans and from the bacterium Pseudomonas mevalonii, in conjunction with sitedirected mutagenesis studies, have revealed details of the mechanism of catalysis. The reaction catalyzed by human HMG-CoA reductase is a target for antihypercholesterolemic drugs (statins), which are intended to lower cholesterol levels in serum. Eukaryotic forms of the enzyme are anchored to the endoplasmic reticulum, whereas the prokaryotic enzymes are soluble. Probably because of its critical role in cellular cholesterol homeostasis, mammalian HMG-CoA reductase is extensively regulated at the transcriptional, translational, and post-translational levels.
3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA)- reductase inhibitors (statins) are mainly considered for long-term use and often constitute part of a multiple-drug regime [2]. Besides common adverse drug effects, such as nausea, abdominal discomfort and headaches, all statins harbour the risk of myopathy and fatal rhabdomyolysis.
The interest in HMG-CoA reductase inhibitors is presently not only due to their role as cholesterol-lowering agents. Instead the fact that mevalonate (the key metabolite whose synthesis is regulated by HMG-CoA reductase) also serves as a precursor for the biosynthesis of non-sterol isoprenoid products essential for initiation of DNA synthesis and cell growth, has attracted increasing attention during the last 10 years.
Atorvastatin (Lipitor) is a member of the drug class known as statins. It is used for lowering cholesterol. Atorvastatin inhibits the rate-determining enzyme located in hepatic tissue that produces mevalonate, a small molecule used in the synthesis of cholesterol and other mevalonate derivatives. This lowers the amount of cholesterol produced which in turn lowers the total amount of LDL cholesterol. Atorvastatin is a competitive inhibitor of HMGCoA reductase.
Atorvastatin under the brand name Lipitor is the largest selling drug in the world. Atorvastatin calcium tablets are currently marketed by Pfizer under the trade name Lipitor. In some countries it is also be sold under the trade names Sortis, Torvast, Torvacard, Totalip, Tulip, Xarator, Atorpic, Liprimar, and Atorlip. Pfizer also packages the drug in combination with other drugs, such as is the case with its Caduet.
Lipitor is a cholesterol-lowering drug. The doctor may prescribe it along with a special diet for high blood cholesterol or triglyceride levels. The drug works by helping to clear harmful Low-Density Lipoprotein (LDL) cholesterol out of the blood and by limiting the body's ability to form new LDL cholesterol [1, 6, 7]. High triglyceride levels lead to Electrical Arrhythmias is originated in the heart's upper chambers, the Atrial Fibrillation (AF or A Fib). More than 2 million people in the United States have atrial fibrillation, making it a very common heart rhythm disorder. In A Fib, the heartbeat is irregular and rapid, sometimes beating as often as 300 times a minute, about four times faster than normal. Although it isn't life threatening, A Fib can lead to other rhythm problems, chronic fatigue and congestive heart failure. Chances of having a stroke are f five times higher for those with A Fib.
Bioinformatics and Cheminformatics software tools like HyperChem, SPDBV (Swiss Protein Data Bank Viewer), GOLD (Genetic Optimization for Ligand Docking) have been utilized for drug design in this paper.
GOLD stands for Genetic Optimization for Ligand Docking. This software uses genetic algorithm to provide docking of flexible ligand and a protein with flexible hydroxyl groups. Otherwise the protein is considered to be rigid. This makes it a good choice when the binding pocket contains amino acids that form hydrogen bonds with the ligand.
The method used for modeling a new drug for heart disorders making 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG CoA reductase) as a target and Atorvastatin, as the drug used (ligand) includes the following steps,
Once potential drugs have been identified by the methods described above, other molecular modeling techniques may then be applied. For example, geometry optimization may be used to "relax" the structures and to identify low energy orientations of drugs in receptor sites. Molecular dynamics may assist in exploring the energy landscape, and free energy simulations can be used to compute the relative binding free energies of a series of putative drugs.
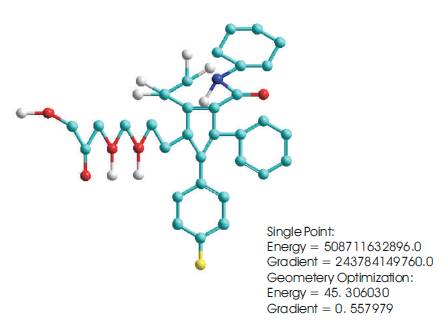
ATORVASTATIN (C33H35FN2O5)
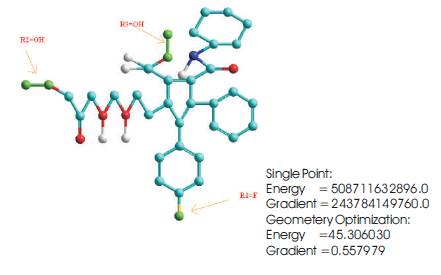
STEP 2 (Replacing R- Groups with Alcoholic Groups)
FIRST MOLECULE (R1=F, R2=OH, R3=OH)
STEP 3: LIGAND-1 (R1=F,R2=OH,R3=OH)
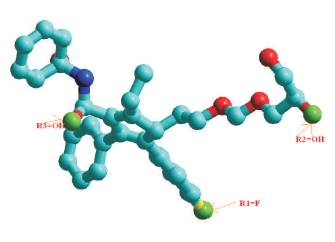
STEP-4 LIGAND:1 (R1=F,R2=OH,R3=OH)

ENERGY= -7526.7823 STEP-5 PROTEIN-1
This is the main method where ligand binds to protein with maximum binding affinity.
Docking is performed using geometrically optimized molecules as ligands and protein HMG COA reductase R as protein, converting their hyperchem forms into PDB format using SPDB viewer. GOLD software is used for this purpose. The name of the ligand and protein files are fed to GOLD, the output yet to come is directed to a particular drive with filename and the program is operated.
The output is obtained with a best ranking docking run number and the fitness energy of that run number is noted down. The binding energy is obtained as:
This is performed for all the 10 ligands:-
Ligand 1
Fitness
S (hb_ext) = 39.92 ; S (vdw_ext) = 5.72
S (hb_int) =0.00 ; S (vdw_int) =-6.02
External energy =39.92+5.72 = 45.64
Internal energy = 0.00+-6.02 = -6.02
The value of 'Y2' is -45.64
The binding affinity is maximum when the binding free energy is minimum i.e., it determines the stable state. The binding free energy of ligand molecules is given in Table 1 which are calculated using the formula,
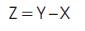
where,
Z – Binding free energy
Y – Energy of the protein ligand optimisation + Fitness Energy
i.e., Y = Y1 + Y2
X – Energy of the optimized ligand devoid of solvation Parameters
The binding free energy of the ligand derivative molecules is obtained by eliminating the ligand energy value from their energy values obtained.
i.e., Zn →Zn+1 = Zn+1 – Zn
The binding free energy for all the novel molecules is obtained by the above calculating formula and is given in Table2.
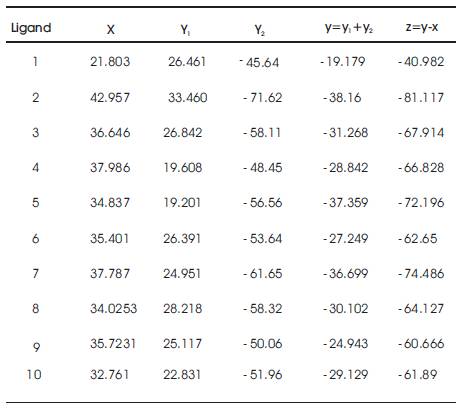
Table 1. Binding Affinity and Binding free energy of Ligand Molecules

Table 2. Binding free energy of Novel Molecules
From the results obtained, the binding free energy of ligand-6 is minimum and therefore the maximum binding affinity is observed for it.
As such molecule 6 is confirmed as the best lead molecule.
One of the most common diseases found among the worlds population has been in steep rise, which is called the cardiovascular disease, which has effected more than one-third of the worlds population. These diseases are found to be an associated one, its being a challenge to the medical field.
Drug designing, one of the hottest topics have found its new path way to create a history in the field of medical science. The lead compound analysis starts with Computer Aided Drug Design (CADD), assisting to identify and to optimize the right compound. The technique helps in generating a suitable compound specific to the disease; thereby an effective treatment is achieved. Molecular modeling method has been used for modeling a new molecule for heart disorders cancer using Atorvastatin, a drug which's already designed. This drug is drawn using hyperchem software, and its R groups are modified by replacing different functional groups like F,OH,Cl,H,Br,CH3 ,CH2OH,I etc in its place. The molecules designed as such are optimized using different algorithms and their affinity is checked with the protein. The binding free energy of the protein is calculated by performing docking process. The molecule with minimum binding energy will have the maximum binding affinity. The binding free energy is calculated by the formula Z = Sum of the energy of optimized ligand devoid of solvation parameters and the energy of the protein-ligand optimization. The binding free energy of the designed molecules is obtained by eliminating the energy of the main molecule i.e. Atrovastatin. From the results obtained its clear that ligand 2 have the maximum binding affinity. So this molecule is determined as the best lead molecules targeting Hmg CoA reductase for curing cardiovascular diseases computationally.